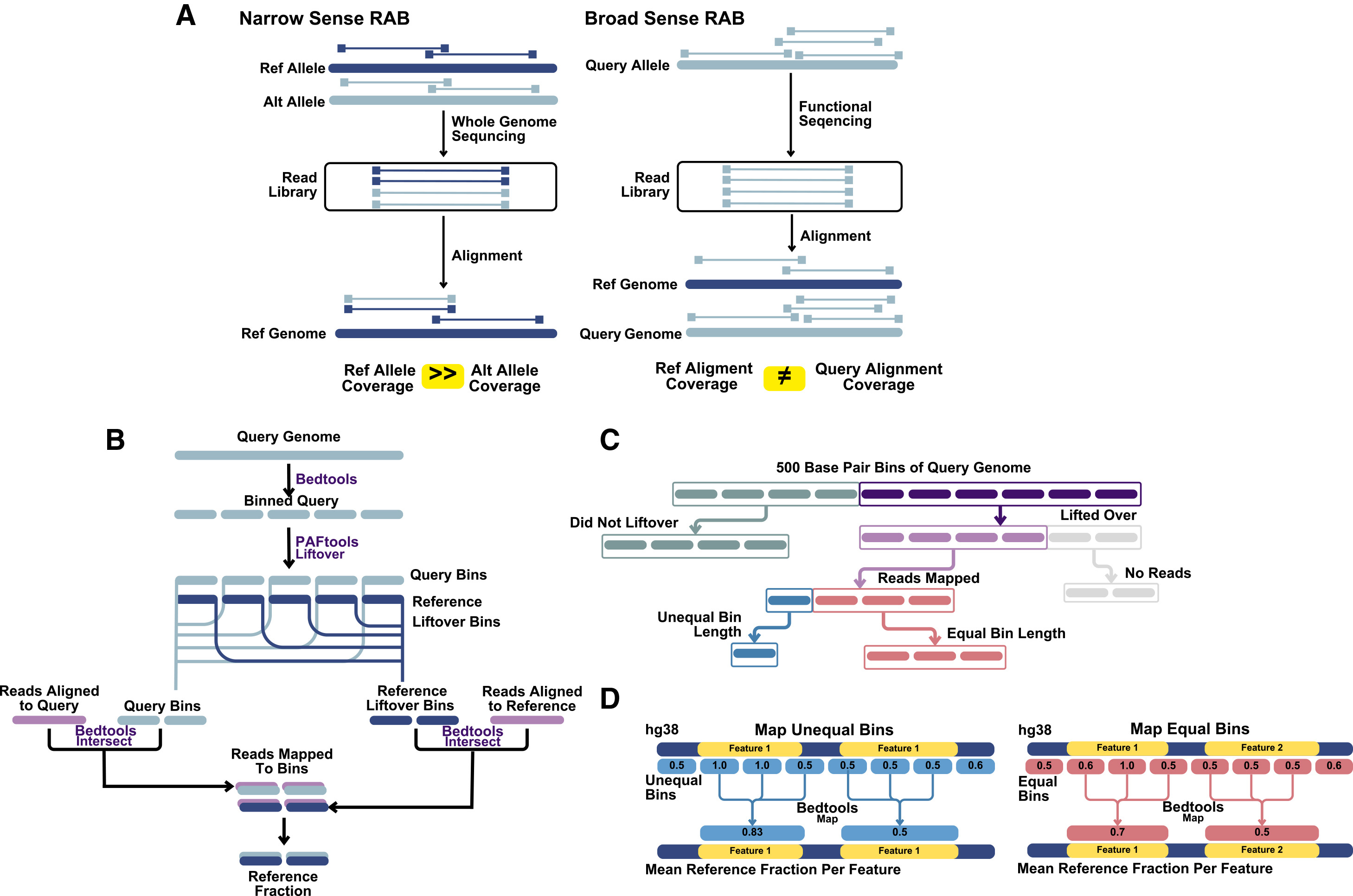
Abstract:
When aligning next-generation sequencing (NGS) reads to a reference genome, differences between the true genome of the individual under study and the reference result in a biased interpretation of aligned data through systematic errors known as reference alignment bias (RAB). The degree to which RAB impacts functional readouts has not been thoroughly quantified. Leveraging resources from the Human Pangenome Reference Consortium, here we quantify RAB in functional genomics assays. Our results indicate that, on average, 0.2% of the genome is susceptible to bias in RNA sequencing (RNA-seq) studies, 1% in ATAC-seq, and 3% in WGBS when using the human reference hg38. Our study quantifies the effect of RAB on functional assays and highlights the importance of using an adequately representative reference genome.


