Abstract:
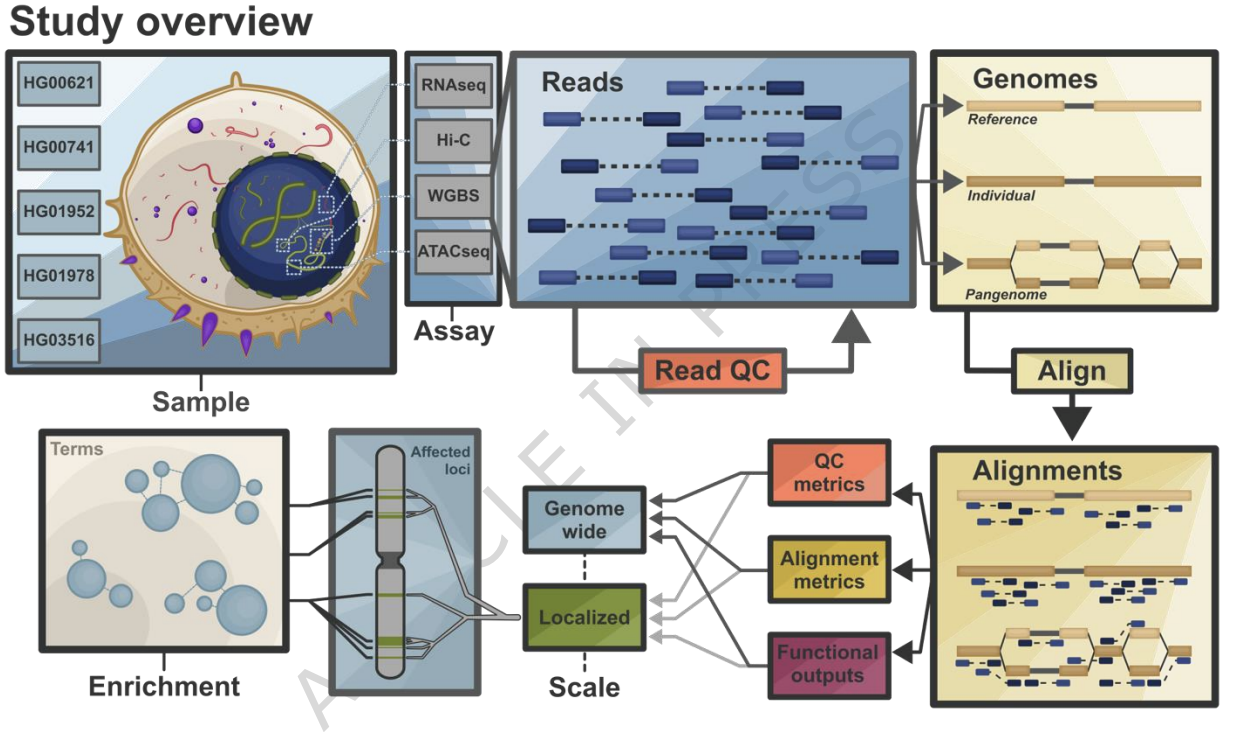
The human genome reference established a shared coordinate system for genome function, but it is incomplete and not fully representative of human diversity. Here, we benchmark how genome representation and corresponding analytical frameworks for each representation shape functional genomics using chromatin accessibility sequencing (ATAC-seq), RNA sequencing, whole-genome bisulfite sequencing, and chromosome conformation capture (Hi-C) data from lymphoblastoid cell lines derived from five individuals with fully phased genome assemblies. We compare results across hg38, CHM13, the draft human pangenome, and each individual’s maternal and paternal assemblies. Because current pipelines and quality control conventions are tuned to hg38, several of these comparisons reflect genome representation in the context of available methods, rather than sequence alone. Individual identity accounts for 57.52-78.47% of total variance in functional estimates, whereas genome choice contributes 0.002-7.85% and sample-by-genome interactions contribute 0.63-5.43%. About 2% of biological signals are detectable only with personal assemblies. Although these effects are modest overall, some biologically important features remain inaccessible to linear references. Consistent with this, graph-based DNA methylation analysis in the human pangenome reveals a non-reference AluY5a insertion within a putative TNKS enhancer at chromosome 8p23.1 that becomes visible and hypermethylated only in the pangenome.


