Abstract:
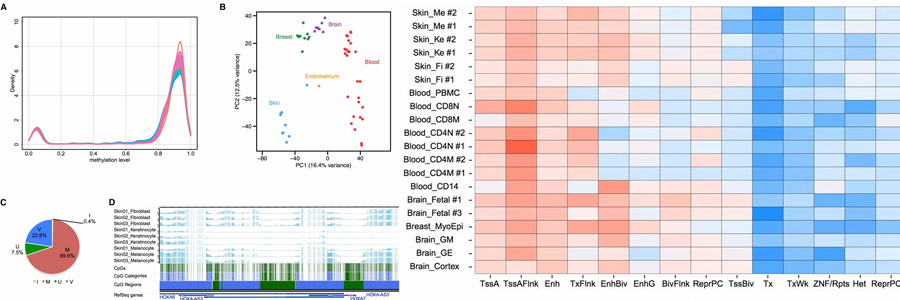
DNA methylation is an important epigenetic modification involved in many biological processes and diseases. Many studies have mapped DNA methylation changes associated with embryogenesis, cell differentiation, and cancer at a genome-wide scale. Our understanding of genome-wide DNA methylation changes in a developmental or disease-related context has been steadily growing. However, the investigation of which CpGs are variably methylated in different normal cell or tissue types is still limited. Here, we present an in-depth analysis of 54 single-CpG-resolution DNA methylomes of normal human cell types by integrating high-throughput sequencing-based methylation data. We found that the ratio of methylated to unmethylated CpGs is relatively constant regardless of cell type. However, which CpGs made up the unmethylated complement was cell-type specific. We categorized the 26,000,000 human autosomal CpGs based on their methylation levels across multiple cell types to identify variably methylated CpGs and found that 22.6% exhibited variable DNA methylation. These variably methylated CpGs formed 660,000 variably methylated regions (VMRs), encompassing 11% of the genome. By integrating a multitude of genomic data, we found that VMRs enrich for histone modifications indicative of enhancers, suggesting their role as regulatory elements marking cell type specificity. VMRs enriched for transcription factor binding sites in a tissue-dependent manner. Importantly, they enriched for GWAS variants, suggesting that VMRs could potentially be implicated in disease and complex traits. Taken together, our results highlight the link between CpG methylation variation, genetic variation, and disease risk for many human cell types.


